.png)
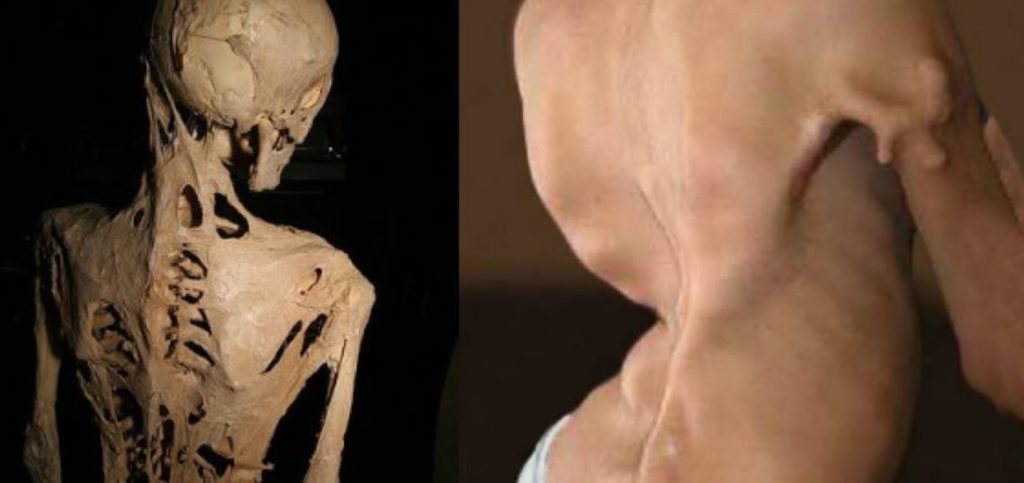
تخيل أن جسدك بكل ليونته وحركته يبدأ ببطء في التحول إلى كتلة صلبة من العظام، أو أن عضلة كتفك، أو رباط ركبتك، أو حتى أنسجة فمك، تتحول تدريجيا إلى عظم لا يمكن كسره أو إزالته، هذه ليست صورة من فيلم خيالي بل حقيقة قاسية يعيشها من يعانون من متلازمة نادرة تسمى تليف العضلات المعظم المترقي (Fibrodysplasia Ossificans Progressiva – FOP)، والتي تعرف عالميا باسم متلازمة الرجل الحجري.
المرض نادر للغاية فوفق التقديرات يصيب شخصا واحدا بين كل مليوني إنسان تقريبا، لكن الدراسات الحديثة تظهر أن هذا الرقم قد يكون أقل من الواقع، مع وجود مرضى لم يشخصوا بعد بسبب ندرة المعرفة بالمرض وتأخر التشخيص في كثير من الدول، خاصة في أفريقيا وآسيا.
وبالتالي فالنتيجة مأساوية تتمثل في وجود آلاف الأشخاص يعيشون مع هذا المرض دون أن يعرفوا اسمه أو يحصلوا على الدعم المناسب.
لم يفك اللغز الجيني للمرض إلا قبل سنوات قليلة، والسبب هو طفرة محددة في جين يسمى ACVR1، المسؤول عن مستقبل إشارات مهم في نمو العظام.. الطفرة الأكثر شيوعا تعرف ب R206H وهي تجعل المستقبل يتصرف بشكل خاطئ، حيث يستجيب لبروتين يسمى Activin A بطريقة غير طبيعية.
في الوضع الطبيعي هذا البروتين لا علاقة له ببناء العظام، لكنه في أجساد مرضى FOP يطلق سلسلة من الإشارات التي تحول الأنسجة الرخوة إلى عظام، وما يزيد الطين بلة أن هذه العملية غالبا ما تبدأ بعد إصابات صغيرة جدا أو حتى التهابات فيروسية، حيث مجرد سقوط بسيط أو وخزة إبرة قد تؤدي إلى اشتعال المرض وظهور بؤرة جديدة من العظم في مكان لم يكن من المفترض أن يوجد فيه.
وللأسف لا تتوفر إحصائيات محددة لمتلازمة الرجل المتحجر في العالم العربي تحديًا، ولكن يمكن القول إن هذه تصيب شخصا واحدا من كل 2 مليون نسمة تقريبا، ولا يوجد لها ميل عرقي أو جغرافي محدد، مما يعني أنها يمكن أن تحدث في أي مكان في العالم، بما في ذلك العالم العربي.
بداية الرحلة من أصابع القدم إلى الجمود الكامل
يولد معظم المصابين بعلامة واضحة يمكن أن تساعد في التشخيص المبكر، تتمثل في تشوه خلقي في أصابع القدم الكبيرة لكن ما لم يلتفت إليه الأطباء، أن الأعراض تبدأ في الطفولة المبكرة.
أولا تظهر تورمات مؤلمة تعرف ب الاشتعالات، سرعان ما تتحول إلى عظم صلب يبدأ التعظم من الرقبة والكتفين وينزل تدريجيا إلى الصدر والأطراف، وبمرور السنوات يفقد المريض القدرة على تحريك مفاصله واحدا تلو الآخر.
في سن الثلاثين تقريبا يصبح كثير من المرضى مقعدين بالكامل، وأحيانا عاجزين حتى عن فتح أفواههم لتناول الطعام أو الكلام، والمشكلة الأخطر أن العظام الزائدة حول القفص الصدري تعيق التنفس، مما يؤدي إلى قصور رئوي أو التهابات قاتلة، ومتوسط عمر المرضى يتراوح بين 40 و50 عاما فقط.
حتى لا يتحول العلاج إلى كارثة
واحدة من أعظم مآسي FOP ليست في المرض ذاته بل في الجهل به، وأكثر من نصف المرضى يشخصون خطأ في البداية فكثير منهم يعتقد أنهم مصابون بالسرطان، لأن الكتل التي تظهر تحت الجلد تشبه الأورام.
هذا التشخيص الخاطئ يقود الأطباء إلى إجراءات خطيرة مثل الخزعة الجراحية، وهي كارثة بالنسبة لمريض FOP فمجرد جرح صغير يطلق سلسلة من الالتهابات تؤدي إلى تعظم سريع ودائم في موقع العملية.
تشير الإحصاءات إلى أن نحو 67% من المرضى تعرضوا لتدخلات جراحية غير ضرورية، وأن ما يقرب من نصفهم فقدوا الحركة بشكل دائم بسبب هذه الإجراءات.
رحلة البحث عن العلاج
لوقت طويل لم يكن أمام الأطباء سوى تقديم رعاية داعمة، ومحاولة تخفيف الألم باستخدام مضادات الالتهاب أو الكورتيزون، لكن الفهم الجيني للمرض غير المعادلة.
البالوفاروتين (Palovarotene) أول دواء تمت الموافقة عليه في عدة دول، يعمل على منع تكوين الغضروف الذي يسبق نمو العظام الزائدة، لكنه يحمل خطرا كبيرا للأطفال لأنه قد يوقف نمو العظام الطبيعية.
الجاريتوسماب (Garetosmab) جسم مضاد يستهدف بروتين Activin A نفسه، ورغم نتائجه الواعدة، لا يزال في طور التجارب بسبب مخاوف تتعلق بالسلامة.
أما المستقبل فيتجه نحو العلاج الجيني عبر تقنيات مثل CRISPR لتصحيح الطفرة نفسها، أو RNAi لإسكات الجين المعيب. لكن إدخال هذه العلاجات يواجه عقبة كبرى.. أي حقن أو تدخل جراحي قد يشعل المرض ما يجعل طرق التوصيل الجيني خطرا مضاعفا.

كارلي هنروتاي.. إرادة تقاوم التصلب
ومن بين قصص المصابين بهذا المرض، تأتي قصة كارلي هنروتاي التي لم يكن طريقها سهلا، فقد بدأ جسدها بالتحول ببطء إلى شكل حجري، لا يمكنها أن ترفع يدها فوق رأسها أو تنحني، أو تقف على قدميها بشكل طبيعي، اضطرت للاعتماد على كرسي متحرك، وأصبحت حركة فمها محدودة جداً لا تتجاوز بضعة ملليمترات، مما جعل الكلام والأكل مهمة صعبة.
وبالرغم من تشخيص إصابتها بهذا المرض، إلا أنها عاشت طفولة طبيعية نسبيا، وشاركت في ألعاب رياضية مثل السباحة والركض ولعب كرة القدم الأمريكية، قبل أن تحرم من الحركة، في وقت لاحق.
كانت أكبر مخاوفها هي الإصابات، حيث إن أي حادث بسيط يمكن أن يسرع نمو العظام في جسدها، وهو ما يفسر استحالة إجراء أي عملية جراحية لإزالة العظام الزائدة، لأن الجراحة نفسها ستتسبب في نمو إضافي لها.
رغم هذه القيود لم تستسلم كارلي فبعد أن تخلت عن شغفها بكرة القدم، التي كانت تمارسها كطفلة، وجهت طاقتها نحو الدراسة، وتفوقت في جامعتها، رافضةً أن يتم تعريفها بإعاقتها.
المرض لا يقتل الجسد وحده
يفقد المصابون تدريجيا استقلاليتهم، وغالبا ما يصبحون عاجزين عن الحركة في عمر الشباب، ودراسات الجودة الحياتية تظهر أن المراهقين والشباب هم الأكثر تأثرا نفسيا، إذ تتزامن إعاقتهم مع المرحلة العمرية التي يسعى فيها الإنسان للاستقلال وتحقيق ذاته.
الأسر أيضا تدفع ثمنا باهظا نفسيا وماليا فتكاليف المرض ضخمة وغالبها غير مباشر مثل الحاجة إلى تعديلات في المنازل أو صعوبة السفر، إذ تشير الدراسات إلى أن أكثر من 75% من التكلفة الإجمالية تقع على الأسر بشكل غير مباشر.
في مواجهة هذا المرض النادر تلعب المنظمات دورا حيويا، والجمعية الدولية ل FOP (IFOPA)، على سبيل المثال، لا توفر فقط التمويل للأبحاث بل تنشر المعرفة وتنظم حملات توعية لتقليل التشخيص الخاطئ.
برامج مثل Tin Soldiers تعمل على اكتشاف الحالات في مناطق محرومة، حيث غالبا ما يترك المرضى بلا اسم لمرضهم ولا أمل في علاجهم.
صحيح أن متلازمة الرجل الحجري تجسد مأساة الطب الحديث المتمثلة في مرض نادر مدمر،معروف السبب، لكنه عصي على العلاج النهائي حتى الآن.. ومع ذلك فإن التقدم العلمي في فهم الطفرة الجينية وابتكار العلاجات المستهدفة يفتح نافذة أمل جديدة.