

.png)
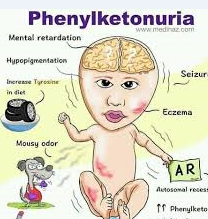
اضطراب وراثي نادر، يصيب الأطفال لكن أعراضه لا تظهر فور الولادة، يسمى الفينيل كيتونوريا (Phenylketonuria – PKU) تكمن خطورته في أن الطفل يبدو سليمًا في أيامه الأولى.
ومع مرور الأسابيع، تبدأ علامات غير واضحة مثل ضعف النمو، والقيء، والرائحة المميزة في البول والعرق، ثم يظهر تأخر في التطور العقلي والحركي إذا لم يُكتشف المرض، ويعزى هذا الاضطراب لخلل في إنزيم مسؤول عن تكسير الحمض الأميني فينيل ألانين الموجود في الأطعمة البروتينية.
كيف يتحول المرض إلى إعاقة؟
وعندما يعجز الجسم عن التعامل مع هذا الحمض، يتراكم في الدم والدماغ إلى مستويات سامة تؤثر مباشرة على خلايا المخ، ما يؤدي إلى تلف عصبي دائم وإعاقة ذهنية إذا لم يُكتشف المرض مبكرًا.
يؤكد الأطباء أن تراكم الفينيل ألانين يؤثر على تطور الدماغ خلال السنوات الأولى من العمر، ما يسبب إعاقة ذهنية متفاوتة الشدة، وصعوبات في التعلم والسلوك، ونوبات صرع، واضطرابات في التركيز والانتباه.
الفحص المبكر.. خط الدفاع الأول
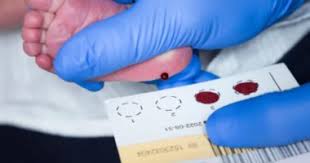
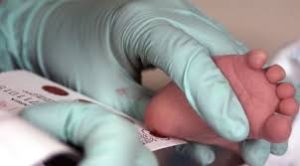
يُعد فحص حديثي الولادة هو السلاح الأهم لمواجهة المرض، حيث يمكن من خلال نقطة دم تُؤخذ من كعب الطفل خلال الأيام الأولى من حياته اكتشاف الإصابة قبل ظهور أي أعراض.
ويؤدي الاكتشاف المبكر إلى تجنّب معظم المضاعفات، إذ يمكن إدارة الحالة بنجاح من خلال نظام غذائي خاص منخفض البروتين يمنع تراكم الفينيل ألانين في الدم.
الحمية الخاصة.. علاج يقي من الإعاقة
لا يوجد علاج نهائي لمرض الفينيل كيتونوريا حتى الآن، لكن الالتزام بالحمية الغذائية منذ الأيام الأولى يُعد كافيًا للحفاظ على نمو طبيعي للطفل.
وتعتمد هذه الحمية على تحديد كمية البروتين الطبيعي في الطعام، مع تناول خلاصات غذائية طبية خاصة تحتوي على الأحماض الأمينية الضرورية باستثناء الفينيل ألانين.
كما ظهرت في السنوات الأخيرة أدوية مساعدة مثل “سابروبتيرين” (Sapropterin) وبيغفالياز” (Pegvaliase)تساعد بعض الحالات على تحسين قدرة الجسم على معالجة الحمض، لكنها لا تغني عن المتابعة الدقيقة والتغذية العلاجية.
مرض وراثي
ينتقل مرض الفينيل كيتونوريا بالوراثة، أي أن الطفل يُصاب فقط إذا ورث الجين المعيب من كلا الأبوين، لذلك تزداد معدلات الإصابة في المجتمعات التي ينتشر فيها زواج الأقارب، إذ ترتفع احتمالات التقاء الجينات الحاملة للمرض في عائلة واحدة.
وفقًا لمراجعة منهجية صدرت عام 2020 بعنوان Global prevalence of classic phenylketonuria… فإن الانتشار العالمي للفينيل كيتونوريا الكلاسيكية هو 6.002 لكل 100,000 مولود حي، أي ما يعادل تقريبًا حالة واحدة من بين كل 16,650 مولود.
أما في دراسة عربية تُغطّي الدول العربية وتركيا وإيران صدرت عام 2018، فوجِد أن انتشار الفينيل كيتونوريا الكلاسيكي ضمن برامج فحص المواليد الوطنية يتراوح ما بين 0.005٪ إلى 0.0167٪، أي بين 1 في كل حوالي 20,000 مولود (الإمارات) إلى 1 في كل حوالي 6,000 مولود (تركيا)، هذه الأرقام تظهر أن بعض الدول العربية تمتلك معدلات قريبة جدًا من أعلى التقديرات العالمية.
التحدي الأكبر.. بين الفقر ونقص الصيغ الطبية
رغم أن اكتشاف المرض مبكرًا يمنع الإعاقة، إلا أن العديد من الأسر العربية تواجه صعوبات في توفير الصيغ الغذائية الطبية الخاصة بسبب ارتفاع أسعارها أو نقصها في بعض المحافظات.
ويؤكد أطباء الأيض أن غياب هذه الصيغ يؤدي إلى ارتفاع مستوى الفينيل ألانين مجددًا، مما يعرّض الطفل لمضاعفات عصبية حتى مع التشخيص المبكر، وتتضاعف المعاناة في المناطق الريفية أو الفقيرة التي تفتقر إلى المتابعة الطبية المتخصصة أو برامج الدعم الغذائي المستمر.
يُعد الفينيل كيتونوريا نموذجًا واضحًا للأمراض التي يمكن الوقاية من إعاقتها عبر وعي المجتمع وفحص المواليد بشكل إلزامي، فالمرض في جوهره ليس حكمًا بالإعاقة، بل قصة يمكن أن تُكتب بنهاية سعيدة إذا توفرت المعرفة والعلاج في الوقت المناسب.
ويبقى التحدي الأكبر في استمرار التوعية ودعم الأسر التي تواجه هذا الاضطراب، حتى لا يتحوّل مرض يمكن السيطرة عليه إلى مأساة تُضاف إلى سجل الإعاقات القابلة للوقاية.
مواد سامة تخترق غشاء المخ
تقدّم الدكتورة منى عبد الرازق، أستاذ الوراثة بالمركز القومي للبحوث بمصر، تحليلًا وافيا لـ”جسور” عن طبيعة المرض وعلاجه قائلة :” إن مرض الفينيل كيتونوريا هو ارتفاع حمض الفينيل الألانين في الدم، وهو أحد الأمراض الوراثية المدرجة تحت أمراض التمثيل الغذائي.
وتؤكد أن المرض الأطفال يصيب حديثي الولادة ويصاحبهم طوال حياتهم، ويصاب الطفل بالفينيل كيتونوريا حين يكون الأب والأم حاملين لهذه الطفرة، وهي عبارة عن نقص لإنزيم معين، هذا الإنزيم هو المسؤول في الجسم عن تحليل حمض الفينيل الأنين، وعند نقصه يتحول هذا الحمض إلى مواد سامة تخترق غشاء المخ وتسبب تلفًا لخلاياه كلما ارتفعت نسبة الحمض.

توضح:” وقت الإصابة بالفينييل كيتونوريا لا يسمح للجسم بالتعامل مع الأطعمة بشكل طبيعي نتيجة نقص إنزيم الفينيل ألانين، لذلك يُمنع المصابون به من تناول البروتين، لأن البروتين يحتوي على 20 حمضًا أمينيًا، منهم 19 حمضًا مسموحًا لمرضى الفينيل كيتونوريا بتناولهم، بينما يحتوي على حمض واحد فقط هو الفينيل ألانين، وهو المادة الممنوعة لأنها تُسبب تلفًا في خلايا المخ للمصابين بالمرض.
وتضيف أستاذ الوراثة أن هذا المرض تم اكتشافه بعد عام 2015، ومنذ ذلك الوقت، في حال ظهور أي من علامات المرض، يتم إجراء التحاليل اللازمة في وزارة الصحة المصرية بتكلفة رمزية، وأن المصابين الذين لم يُكتشف مرضهم قبل هذا التاريخ يعانون من إعاقات ذهنية متفاوتة، وأن المرض ينتج غالبًا عن زواج الأقارب، وينتشر بشكل أكبر في الريف والصعيد، حيث توجد أُسر كاملة مصابة بهذا المرض.
وتؤكد:” لا يوجد علاج نهائي للفينيل كيتونوريا حتى الآن، سوى الاكتشاف المبكر واتباع حمية غذائية صارمة خالية من البروتين أو منخفضة البروتين وفق نسب محددة لكل حالة، لأن دخول البروتين إلى الجسم في حالة الإصابة يُعدّ سمًا للدماغ، ويتسبب مع مرور الوقت في إعاقات ذهنية.
وتضيف: الوعي بالمرض ازداد بشكل كبير في السنوات الأخيرة مقارنة بالماضي، حيث أصبحت الأمهات يتبادلن الوصفات الغذائية الخالية من البروتين لإطعام أطفالهن دون إشعارهم بالحرمان، كما أصبحت الألبان المخصصة لمرضى الفينيل كيتونوريا متوفرة في وزارة الصحة، وهو ما ساهم في تحسين جودة حياة الأطفال المصابين بشكل ملحوظ.
إرتفاع زواج الأقارب
وتشير إلى أن خطورة هذا المرض منذ ظهوره تكمن في كونه مرضًا وراثيًا، ونحن نعاني ارتفاع نسبة زواج الأقارب، بالإضافة إلى أننا نعاني أيضًا ارتفاع نسب المواليد، وعدم الاكتشاف المبكر لهذا المرض ومن ثم علاجه كان يعني إنتاج المزيد من الأجيال ذوي الإعاقة، ومزيدًا من المعاناة لعدد ليس بقليل من الأسر، وبالتالي فهو ضرر لمجتمع بأكمله.
المسحة الشاملة
وتؤكد أستاذ الوراثة بالمركز القومي للبحوث :”أن إدراج الفينيل كيتونوريا في المسحة الشاملة لحديثي الولادة كانت هي الخطوة الأفضل والأصعب على الإطلاق للحد من خطورة هذا المرض، حيث عانينا سنوات قبلها من تأخر اكتشاف المرض وبالتالي تأخر الحالات، وكذلك من عدم توافر اللبن المخصص للحمية التي تناسب المصابين به.
وهذه هي الخطوة الثانية التي قدمتها الدولة لمصابي الفينيل كيتونوريا، وهي توفير حصص الألبان اللازمة للمصابين في وزارة الصحة رغم صعوبة الحصول عليها، ونتمنى قريبًا أن نقوم نحن بتصنيع هذه الألبان بدلًا من استيرادها ضمانًا لعدم نقصها.
وتشير إلى أنه بالرغم من التوعية الكبيرة بمرض الفينيل كيتونوريا وخطورته وإدراجه في مسحة المواليد، إلا أن البعض ممن يلدون خارج المستشفيات ولا يخضع أطفالهم للكشف المبكر لا يدركون خطورة عدم إجراء عينة الكعب لحديثي الولادة، ولذلك فنحن بحاجة لمزيد من التوعية
ووتوضح الدكتورة منى عبد الرازق أن هناك ما يقارب 19 مرضًا مشابهًا للفينيل كيتونوريا من الأمراض الوراثية الناتجة عن زواج الأقارب ما زالت قيد البحث والدراسة”.
















































